Light can do a lot of interesting things. It can power applications such as energy storage and biological imaging. In medicine, it can be used to activate drugs in the body, including ones that treat diabetes, cancer, and nervous system disorders.
To develop light-activated drugs, scientists start with photoswitches, molecules that activate, or “turn on,” when exposed to light—but finding ones that do that efficiently is no easy task.
Now, a team of MIT and Harvard University researchers has developed a machine learning approach that accelerates the process, potentially clearing a shortcut to discovery of light-activated drugs: fast and accurate prediction of how good photoswitches are at switching on.
For example, doctors can turn on an anti-cancer drug with light to localize the effect, targeting where the tumor is. Everywhere else, the drug is off.
“That way, you can minimize side effects by having it not act in other parts of the body,” said Simon Axelrod, lead author of the study, published in June in the journal Nature Communications. Axelrod is a graduate student in Harvard’s Department of Chemical and Chemical Biology and MIT’s Department of Materials Science and Engineering (DMSE).
Designing light-activated drugs does come with challenges. One is principal in drug design: making a drug that blocks the right target, typically a protein associated with a disease, so that it produces a therapeutic effect in someone who has the disease. The other is making sure that the drug can get turned on by light. For example, when light is shined on an area of the body, most of the drug molecules should be activated. This success rate is known as the quantum yield.
Drugs can be made with photoswitches if they produce an acceptable quantum yield—that is, they’re good at switching on when exposed to light. In theory, efficient “photoswitchable” drugs can be found through virtual screening, which involves searching databases of compounds. But screening photoswitches is difficult and costly, involving complex calculations and hours and hours of time.
Simulating the unpredictable
To understand the MIT-Harvard team’s approach to the problem, it’s important to understand what the molecules do when light is shined on them: they change shape.
“The idea basically is that you can have a molecule that can exist in two different shapes. And you can switch between those two shapes with light,” said Axelrod.
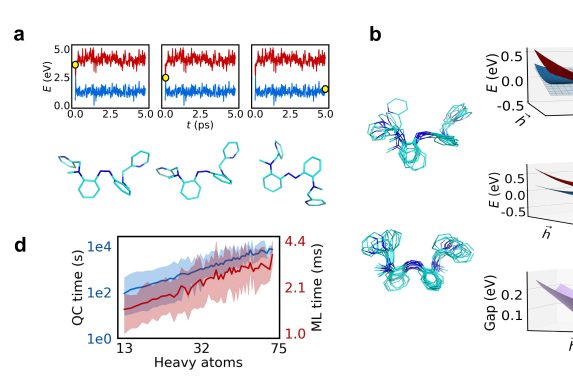
To determine whether molecules changed shape in response to light, Axelrod used atomistic simulation, a computer-aided technique to predict how molecules move in response to their environment. The study focused on azobenzene, a chemical compound that rotates after absorbing light.
“The change in shape comes from each atom inside the molecule moving,” said Axelrod. “In atomistic simulation, we calculate the forces on the atoms and where those forces will push them after a tiny step in time. And then, OK, the atoms are in this new position. Where are they going to be in the next tiny step forward? Do a new calculation; move it a bit. Now where are they going to be in the next step? You get this simulation of how the molecule actually changes its shape in time.”
The problem with predicting what photoswitches will do, though, is they’re inherently hard to predict. When exposed to light—scientists call this their “excited state”—photoswitches exhibit behavior that runs counter to some basic assumptions in molecular dynamics, making it harder to determine which way they might move. There could be hundreds of potential paths, and that means more simulations.
“If you have to do a hundred simulations, it becomes a hundred times more costly,” Axelrod said.
An assist from machine learning
Enter what Axelrod and the researchers call the diabatic artificial neural network, or DANN. It’s a machine learning approach that expands on earlier research involving what’s known as diabatic states. Diabatic states are a mathematical way to describe the forces acting on atoms before and after the molecule absorbs light.
Once you have the diabatic states of a molecule, you can more easily predict how it will move, and ultimately, how efficient it will be at reacting to light. DANN, Axelrod says, is “a very nice, black-box way of making diabatic states that’s generally applicable to even problems that are not our own problem.”
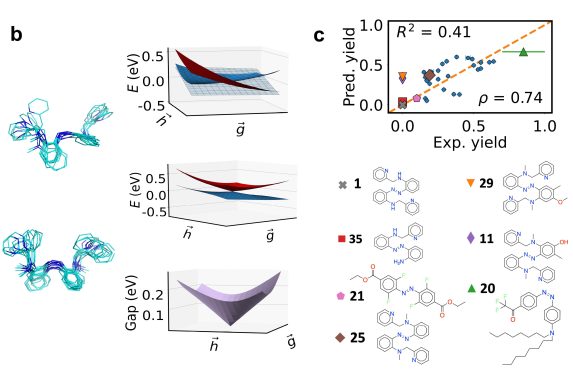
The researchers used the model to screen 3,100 hypothetical molecules—the first large-scale demonstration of such a tool—and identified four potential “hits,” or highly efficient photoswitches, that could, theoretically, be evaluated by pharmaceutical scientists looking to discover new treatments for diseases.
“Folks have used machine learning before to see what happens when a molecule absorbs light. But this is the first time anyone has done it on more than one molecule,” said Professor Rafael Gómez-Bombarelli, the Jeffrey Cheah Career Development Chair in Engineering in DMSE and senior author of the study. “Doing it on thousands of molecules is a step towards really accelerating experiments with computation.”
Yet how good a molecule is at responding to light is just one of many properties that must be fine-tuned for a light-activated drug. Scientists must identify whether the drug binds to the right protein, whether it can be transmitted into the bloodstream or get through the membrane of a cell, and whether it’s safe. It’s a long, slow process.
“They’ll have these molecules, and the ones that bind the right target they’ll then screen for other properties. They’ll spend years fine-tuning these other properties,” said Axelrod. “So the idea of contributing to any of these stages computationally, I would say we hope that we can speed up this process.”
This work was funded by DARPA and the MIT-IBM Watson AI Lab.